第1章
基础介绍
这里会介绍scSpotlight的基础使用. 各文档会有相应的分类Categories和标签Tags, 可以点击查看各分类 对应的文档, 也可以使用左侧菜单栏的搜索框.
这里会介绍scSpotlight的基础使用. 各文档会有相应的分类Categories和标签Tags, 可以点击查看各分类 对应的文档, 也可以使用左侧菜单栏的搜索框.
用户可以从使用devtools 或者 pak 从GitHub上安装,
或者直接使用r-universe作为安装源. 我们推荐使用pak作为安装工具, 速度
相对传统方式快很多, 两者都需要先从CRAN上安装相应的package.
# Install scSpotlight in R:
install.packages('scSpotlight', repos = c('https://obenno.r-universe.dev', 'https://cloud.r-project.org'))# install.packages('pak')
pak::repo_add(scSpotlight = "https://obenno.r-universe.dev")
pak::pkg_install("scSpotlight")# install.packages("devtools")
devtools::install_github("obenno/scSpotlight@v0.0.3")# install.packages("pak")
pak::pkg_install("obenno/scSpotlight@v0.0.3")用户如果有安装docker, 也可以选择使用我们准备好的docker image. 拉取image使用如下命令:
docker pull registry-intl.cn-hangzhou.aliyuncs.com/thunderbio/scspotlight:0.0.3在特定的端口(如8081端口)运行app, 请使用如下命令:
docker run -p 8081:8081 registry-intl.cn-hangzhou.aliyuncs.com/thunderbio/scspotlight:0.0.3 Rscript -e 'scSpotlight::run_app(options = list(port=8081, host="0.0.0.0", launch.browser = FALSE), runningMode="processing")'用户在R console中可以使用如下命令启动app, 然后在浏览器中访问网址127.0.0.1:8081使用app.
scSpotlight::run_app(options = list(port = 8081, host = "0.0.0.0"))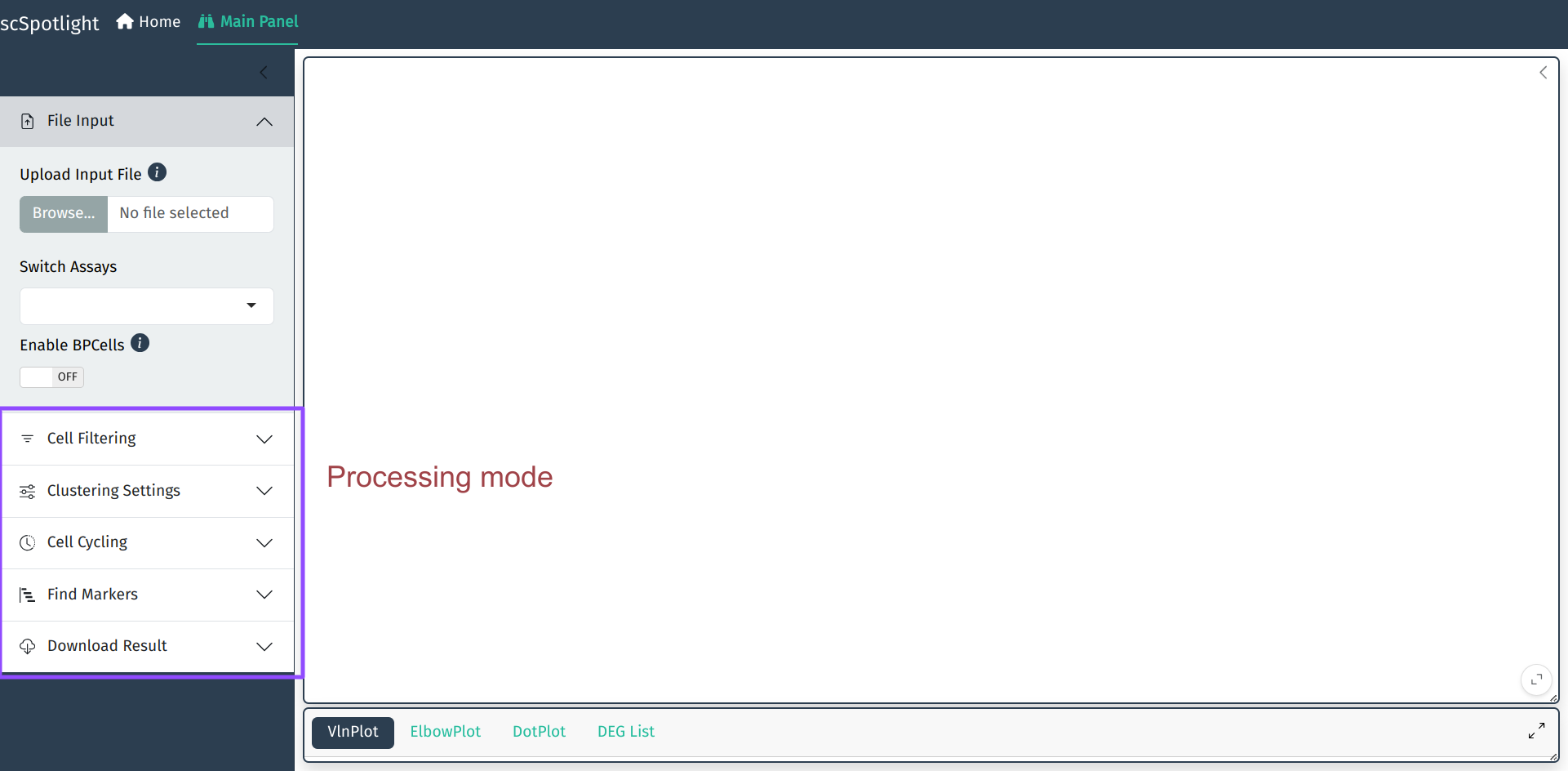
scSpotlight的默认启动模式是processing. 用户可以切换到viewer模式, 仅仅作分群的展示和查看
基因表达.
scSpotlight::run_app(options = list(port = 8081, host = "0.0.0.0"), runningMode = "viewer")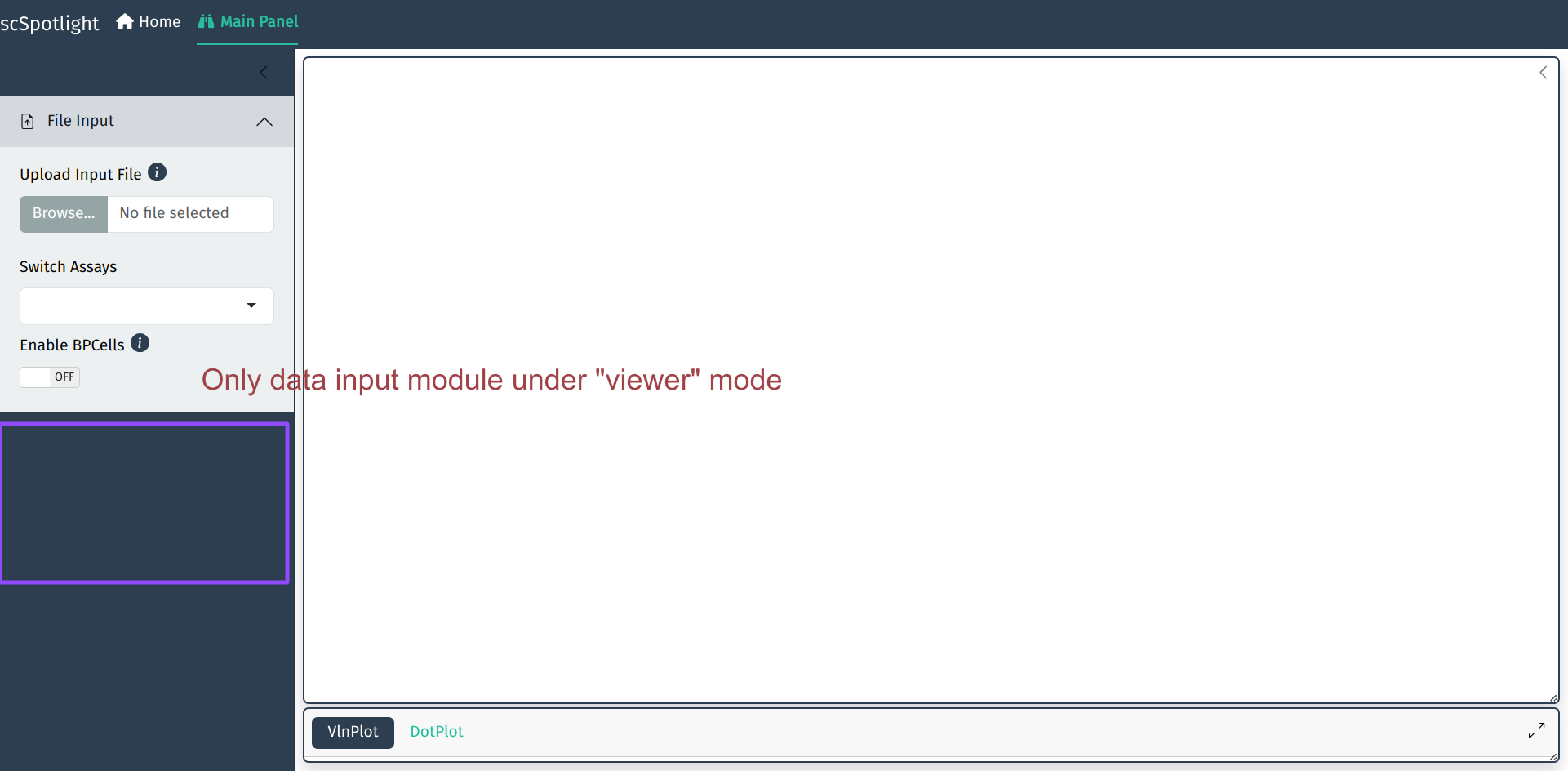
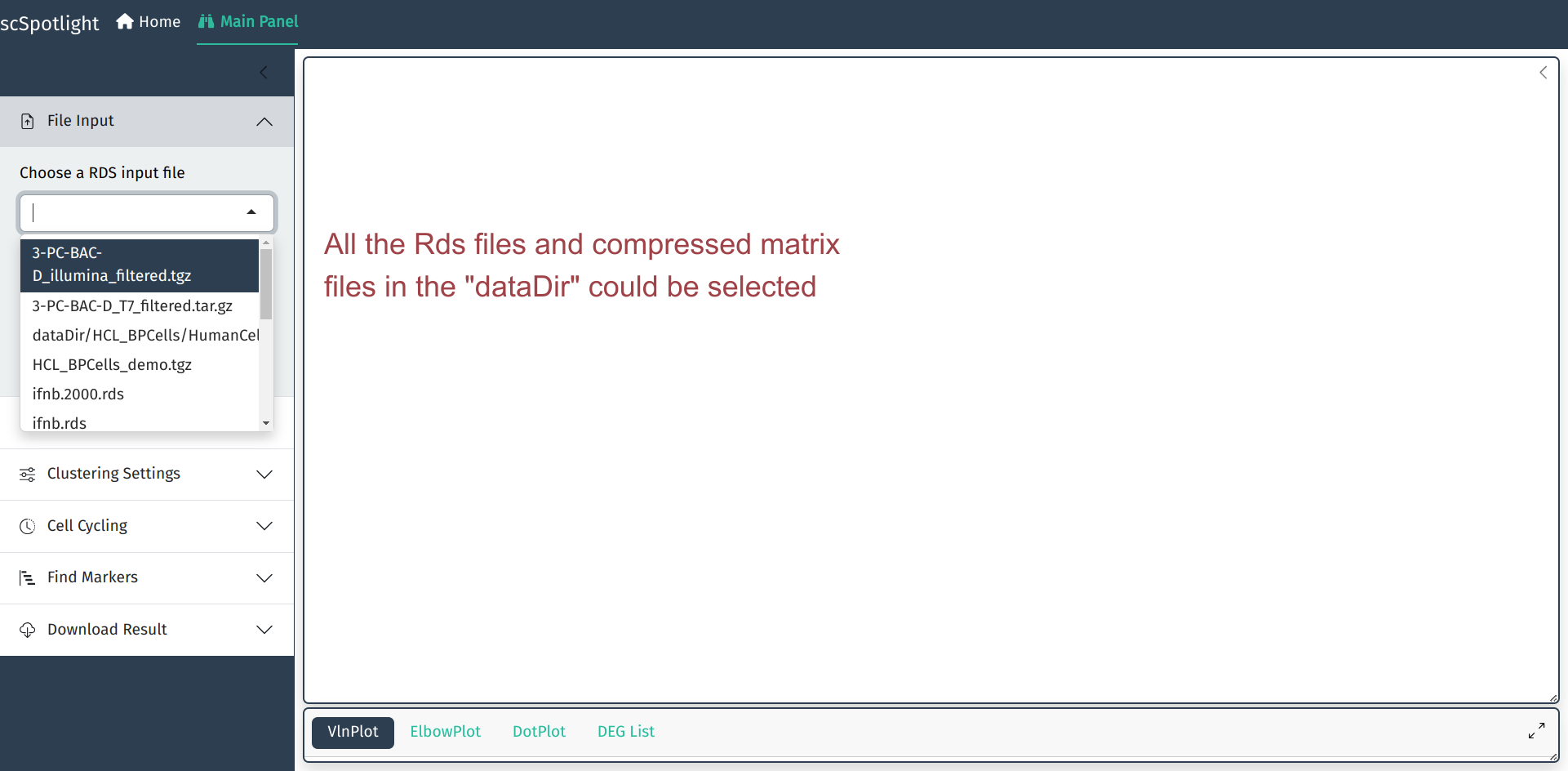
如果载人的数据较大, 可以使用dataDir参数指定存放数据的位置. 此时data loading部分会变成下拉菜单,
用户可以从菜单选取需要载入的Rds文件.
scSpotlight::run_app(options = list(port = 8081, host = "0.0.0.0"), runningMode = "viewer", dataDir = "/path/to/data_directory")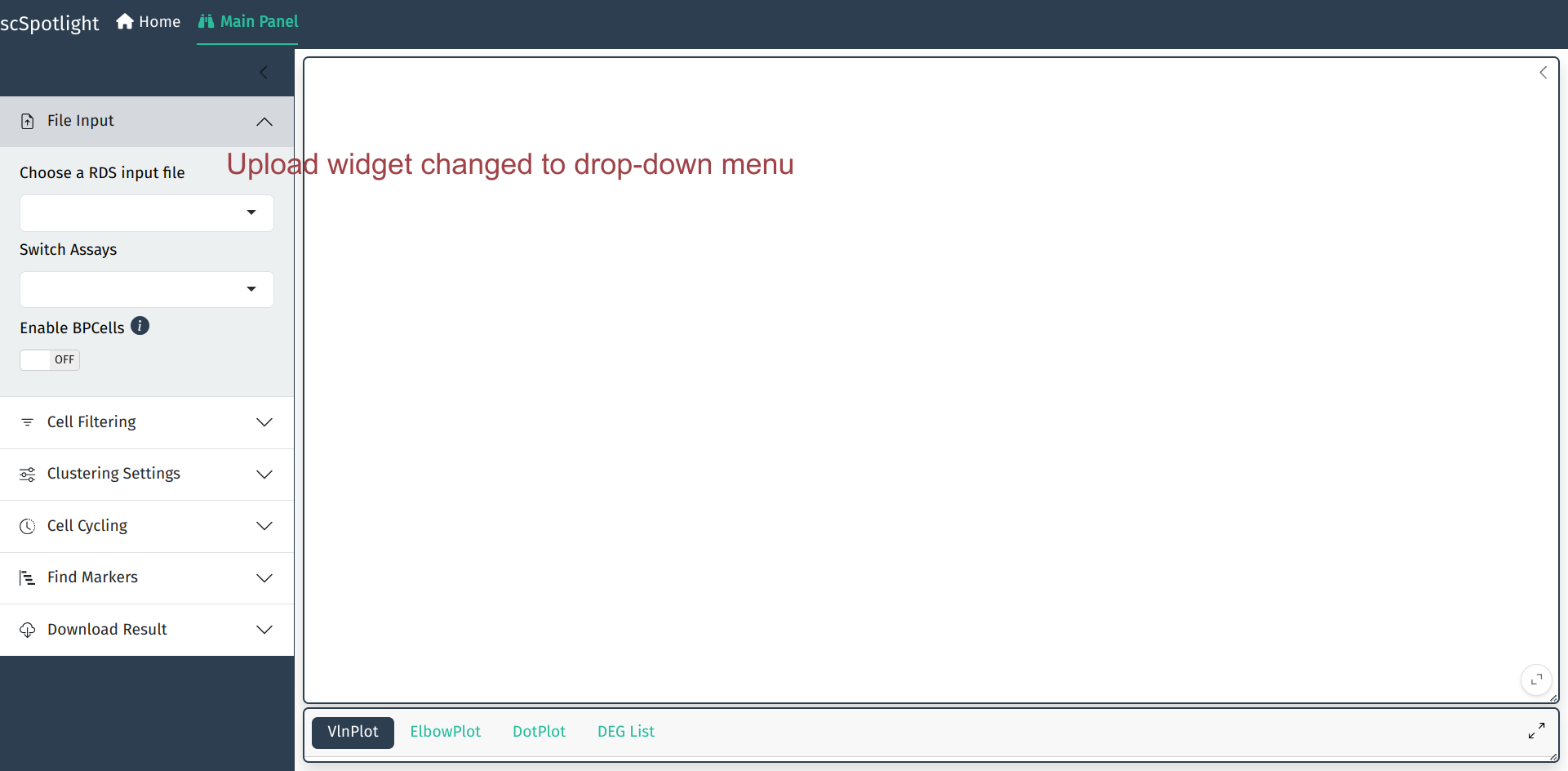
启动APP后, scSpotlight的主面板如下, 左侧边栏包括了数据处理相关功能, 右侧边栏 为分群结果展示的交互选项.
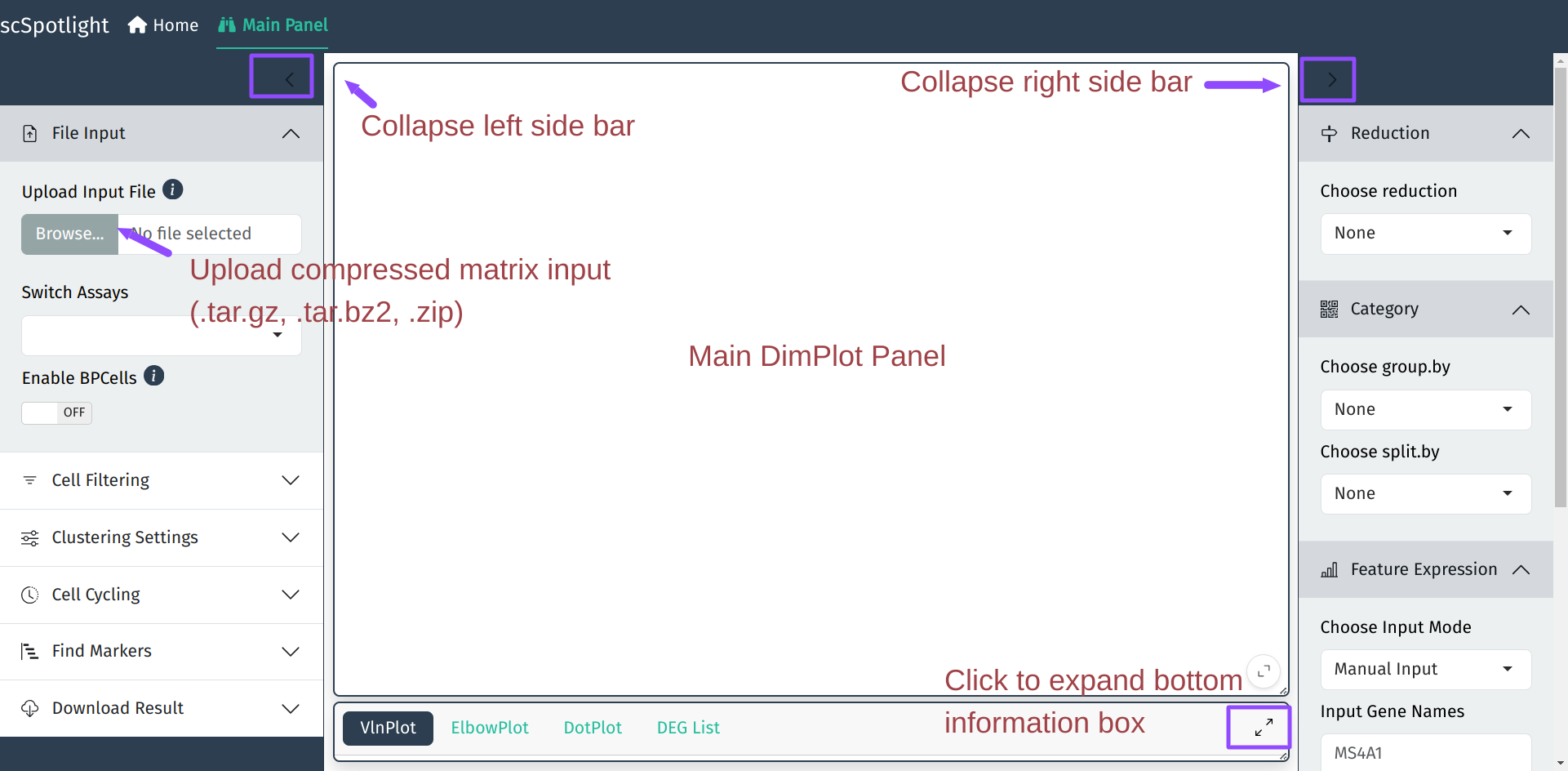
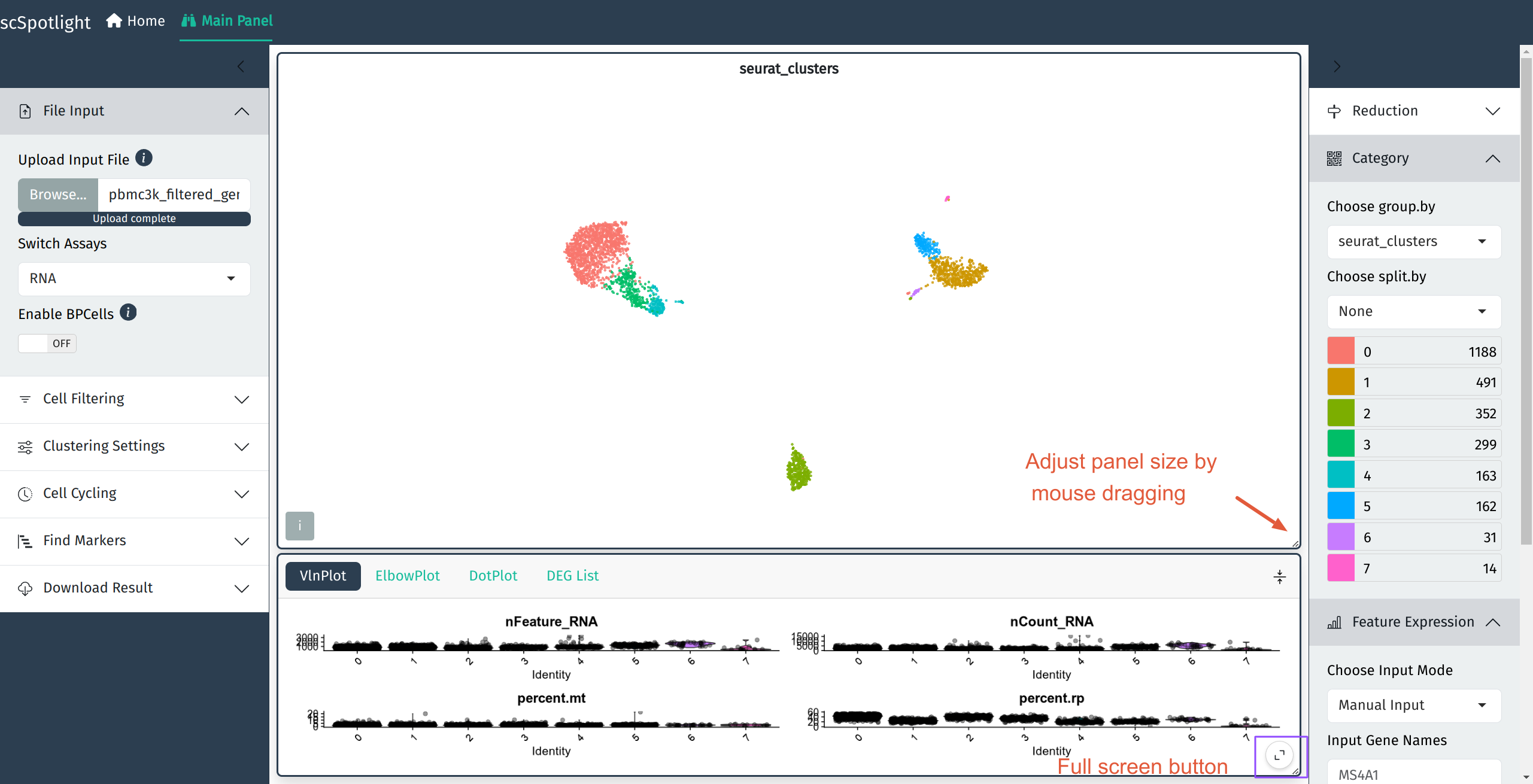
如果使用压缩的矩阵文件作为输入, scSpotlight会自动使用未过滤的细胞和基因 执行"标准化"的分析, 并展示降维和无监督分群的结果. 同时数据集中每个细胞 的基因数(nFeature_RNA), UMI数(nCount_RNA), 线粒体UMI比例(percent.mt) 以及核糖体UMI比例(percent.rp)会统计成小提琴图, 呈现在下方的信息面板内.
展开信息面板后, 用户可以点击右下角的全屏按钮全屏展示也可以使用鼠标拖拽 主面板和信息面板右下角的小三角调整面板大小.
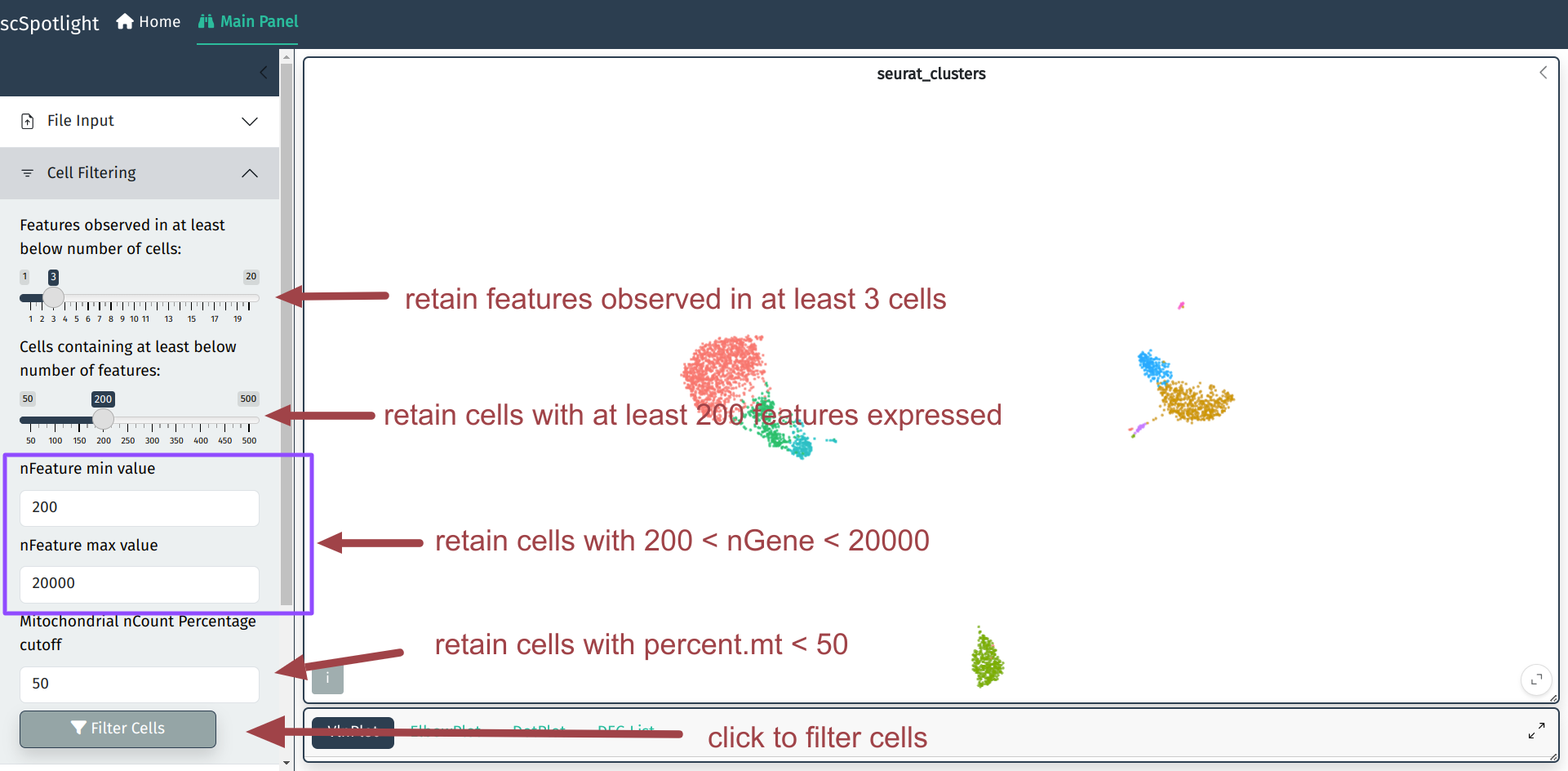
低质量细胞过滤的具体过程请参考Seurat的分析文档, 这里保持和Seurat的标准流程一致, 过滤的内容主要包括如下:
用户可以在细胞过滤模块调整过滤的标准, 根据数据集通常需要调整最大基因数和 线粒体UMI比例的cutoff.
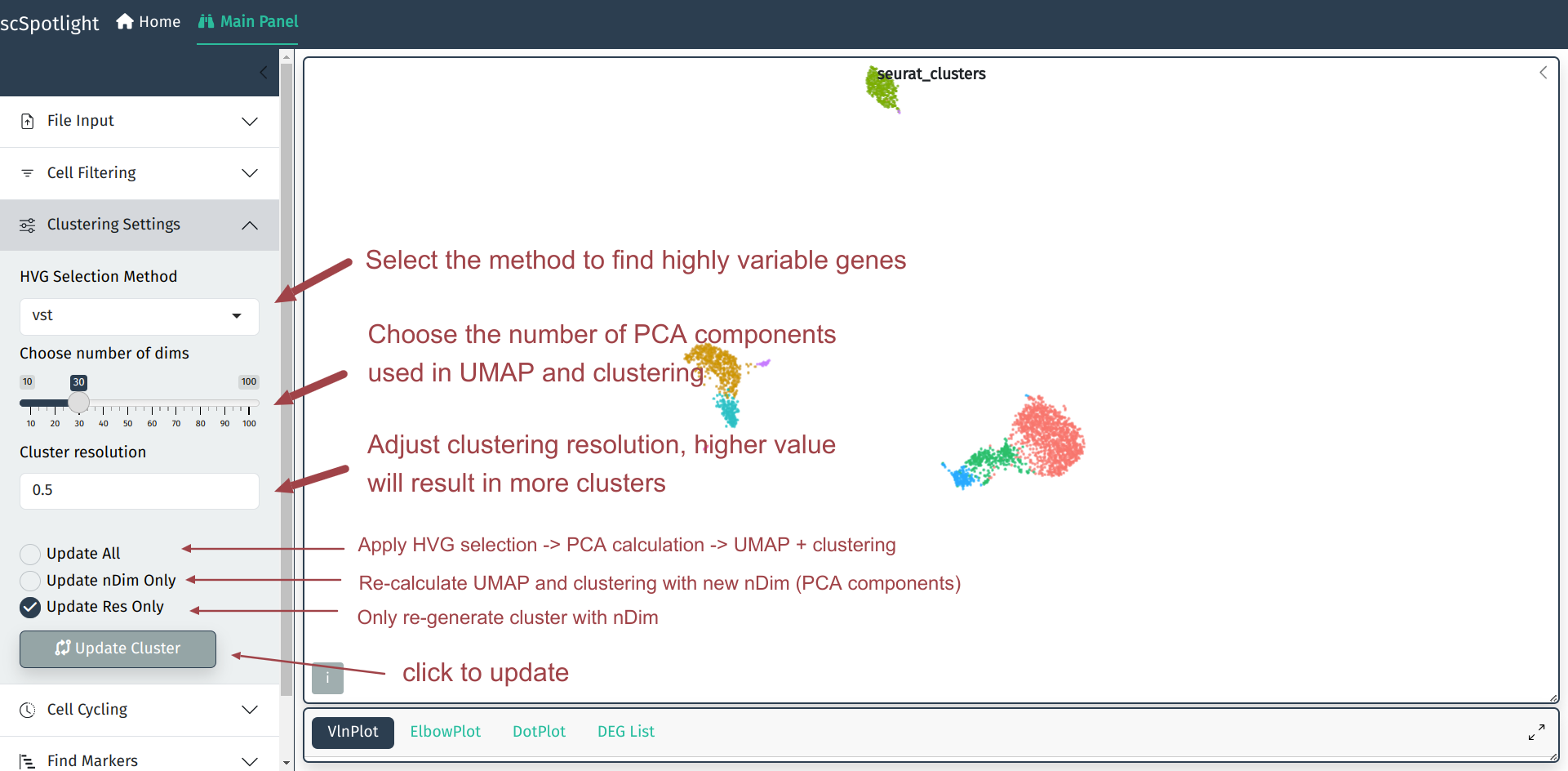
在单细胞分析中默认参数得到的分群结果有时并不能完全满足用户的需求, scSpotlight可以协助用户根据分群结果方便的调整分群参数. 用户可以调整 寻找高可变基因的方法, UMAP和无监督分群中使用到的PCA成分数量, 以及 无监督分群的分辨率. 分辨率越高时得到的cluster数量越多, 用户常常需要 根据不同的数据集和研究的生物学问题调整分群的参数, 找到稀有的细胞类群.
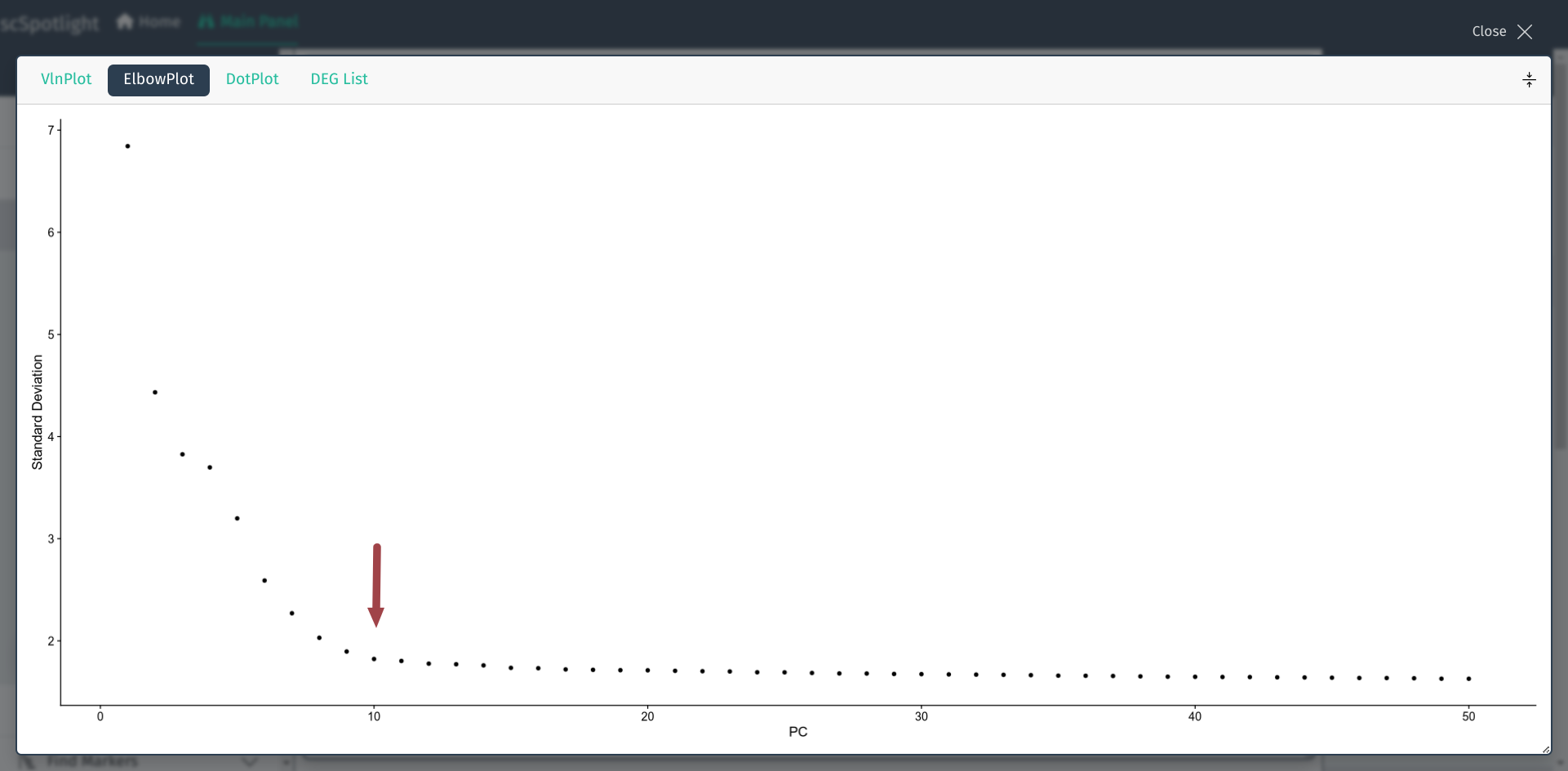
nDim的选择并没有绝对的默认值, 常常根据每个PCA成分对数据变化的"贡献度" 来决定, Seurat流程中常使用肘形图, 用户可以展开面板查看, y轴是PCA成分可以解释的数据变化比例, x轴是根据贡献度排序的成分值, 选择"拐点"的成分值即可. 例如Seurat pbmc3k教程中使用了nDim=10.
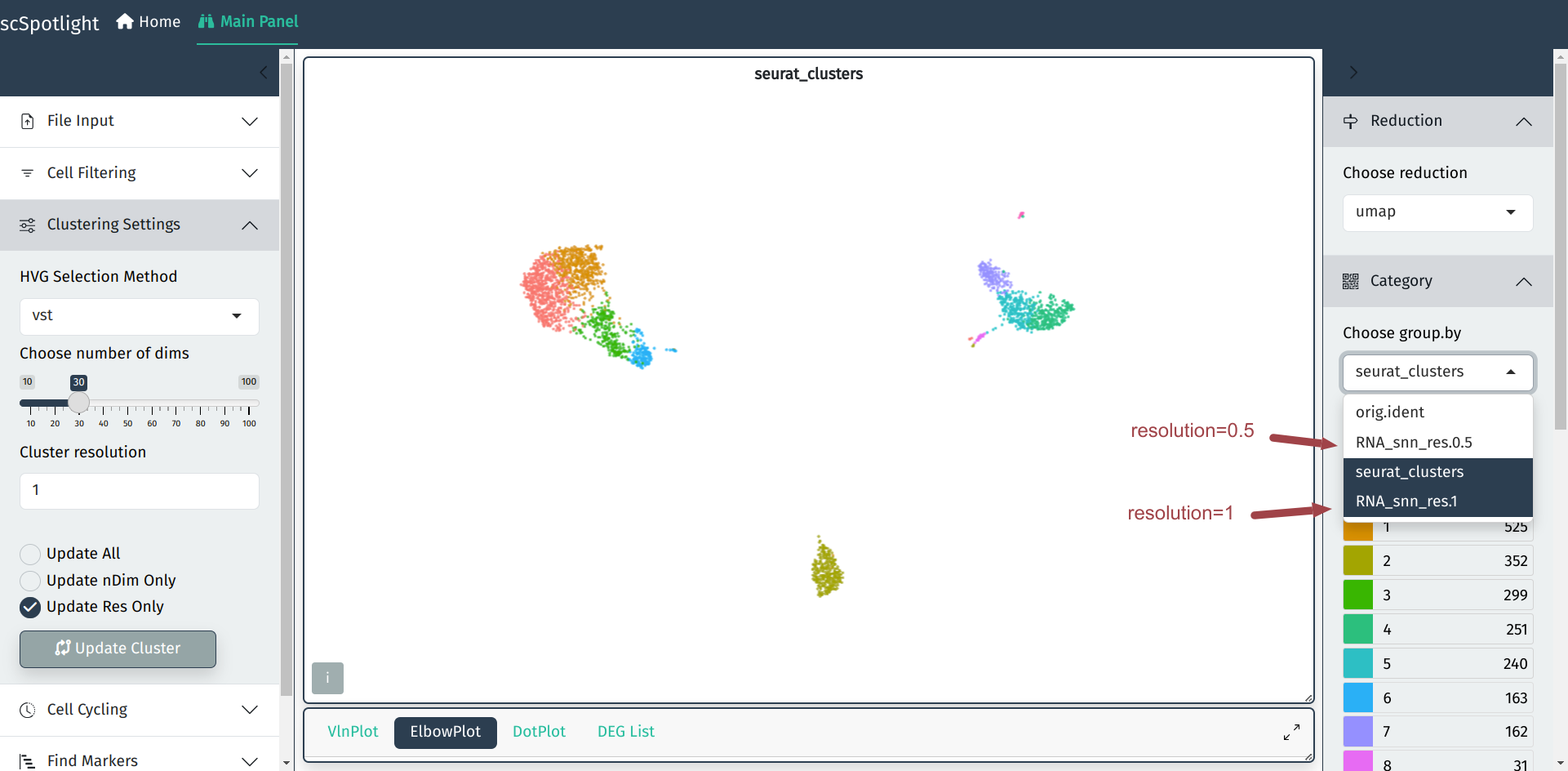
更新分辨率后, 不同的分辨率的结果可以通过在右侧切换分组信息查看.
完成了细胞分群后, 单细胞分析中最频繁的需求应该就是查看基因表达了. scSpotlight 支持同时查看多个基因的Seurat::FeaturePlot, 也支持交互式展示的单个基因表达情况.
用户在右侧边栏Feature Expression模块可以输入多个基因的名字(symbol), 主界面 的细胞分群图会自动更新为这些基因表达的FeaturePlot.
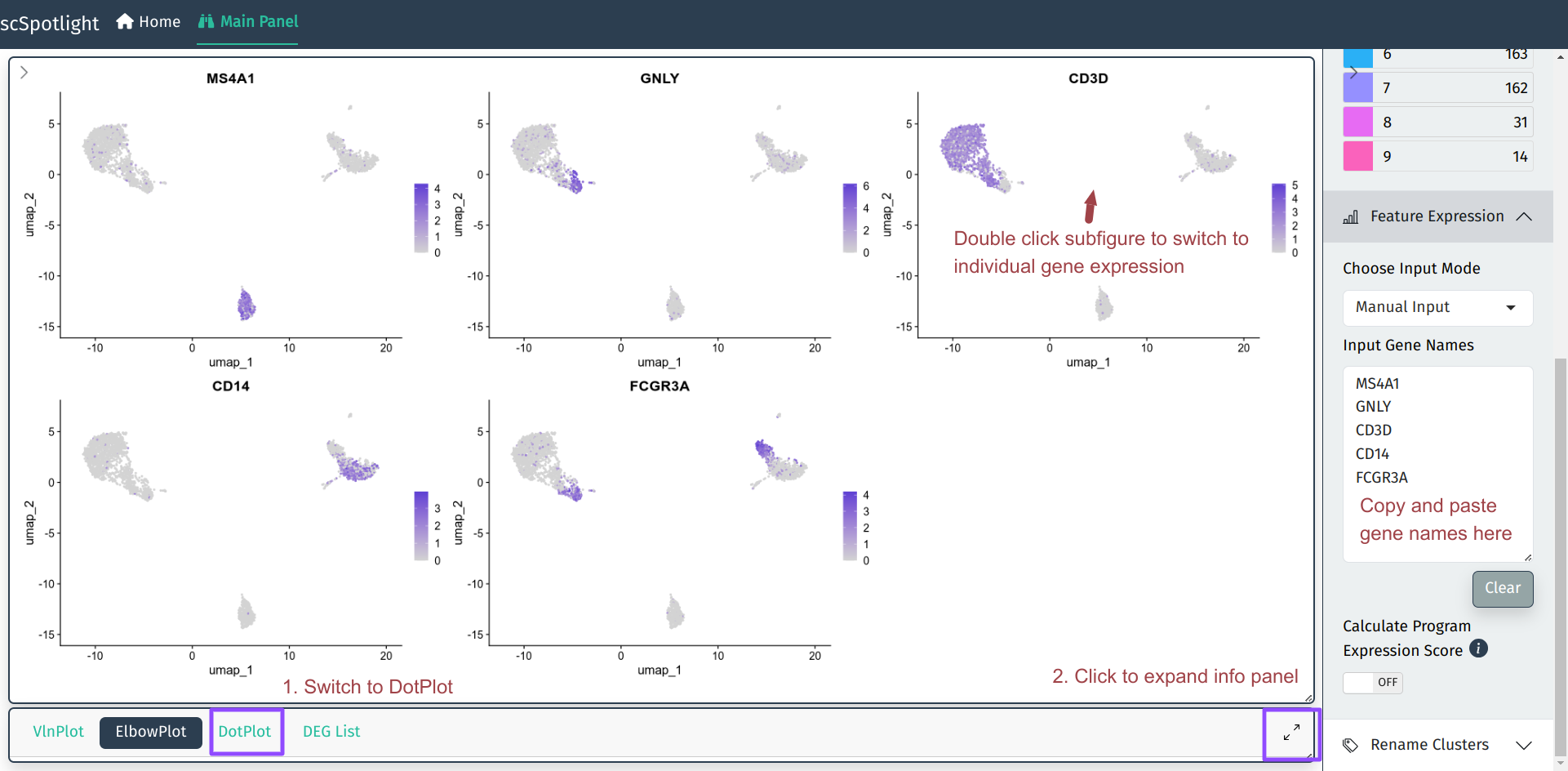
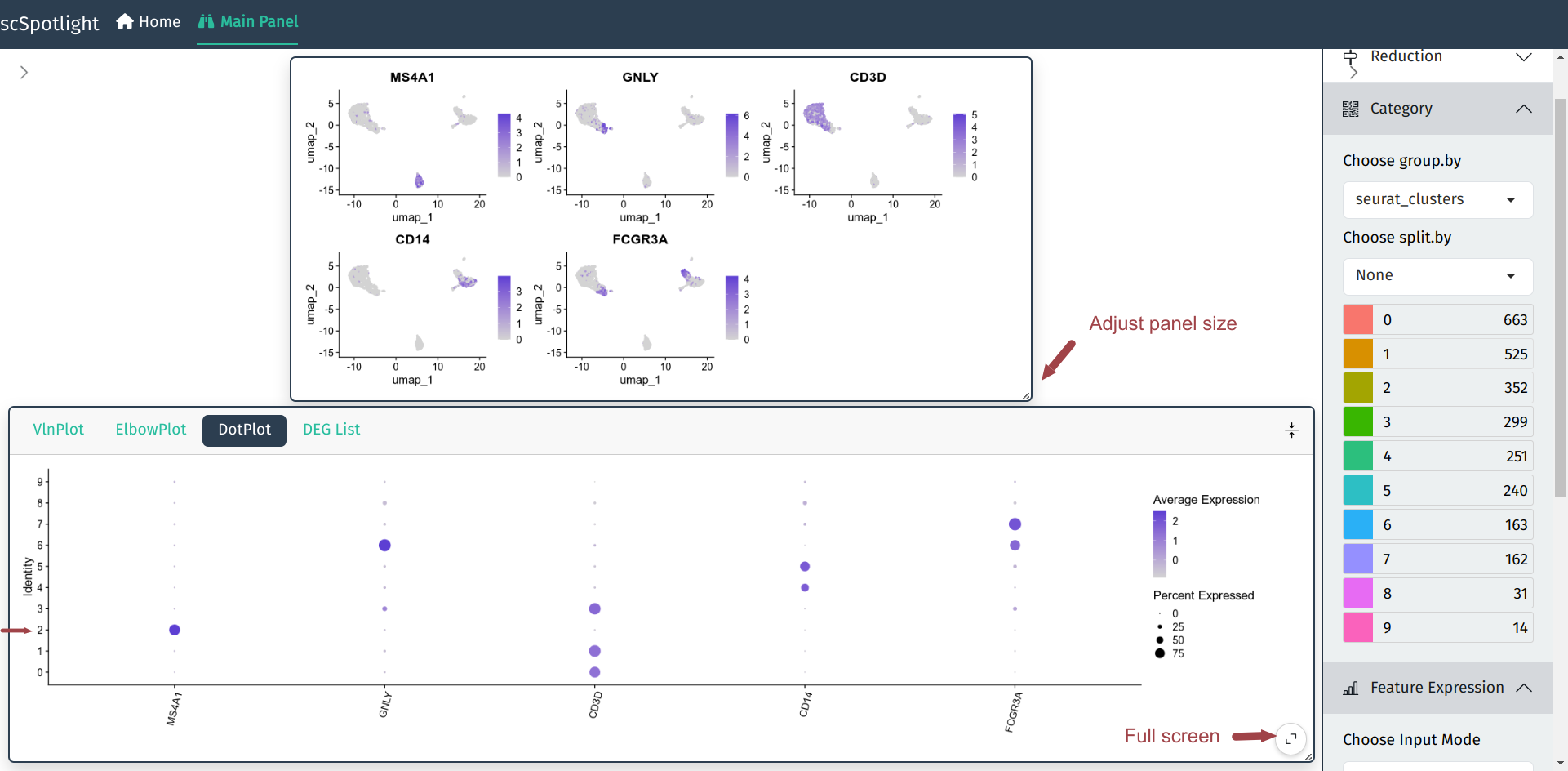
用户可以展开信息栏查看输入基因的Seurat::DotPlot结果, 可以对照FeaturePlot和DotPlot 确认每个cluster的细胞类型, 例如下图中的cluster 2特异表达MS4A1 (CD20), 可以确认为 B cell.
用户在FeaturePlot中双击感兴趣的基因可以切换为单基因的表达. 类似Dotplot, 对于单个基因 可以在信息面板中查看这个基因的表达的小提琴图(Seurat::VlnPlot), 方便直观的确认这个 基因在各个cluster中的表达情况.
通常的单细胞分析中会基于无监督分群的结果选择有相应marker表达的分群 注释为对应的细胞类型, 如特异表达CD3D的为T cell. 实际应用过程中 无监督分群的结果有时并不完美, 需要手动选择细胞进行注释. scSpotlight允许 用户使用两种方案选择感兴趣的细胞.
在右侧边栏的Category模块选择细胞分群信息(如seurat_cluster)后, 用户可以从Rename Cluster模块的Select Cells from Category下拉 菜单中选择相应的细胞分群(如选择1, 2, 对应了seurat_cluster中的cluster1和cluster2). 选择后右下脚会出现提示反映所选择的细胞数量. 用户可以在New Category Name 中输入需要新增的分组信息名称(如celltype), 在Assign As中输入所选 细胞对应的细胞类型(如 T cell), 那么所选细胞就会在celltype分组中被 注释为T cell, 其他的细胞注释为unknown.
用户可以按住shift键启用套索工具选择感兴趣的细胞, 然后使用和上述一样的 方法注释所选细胞.