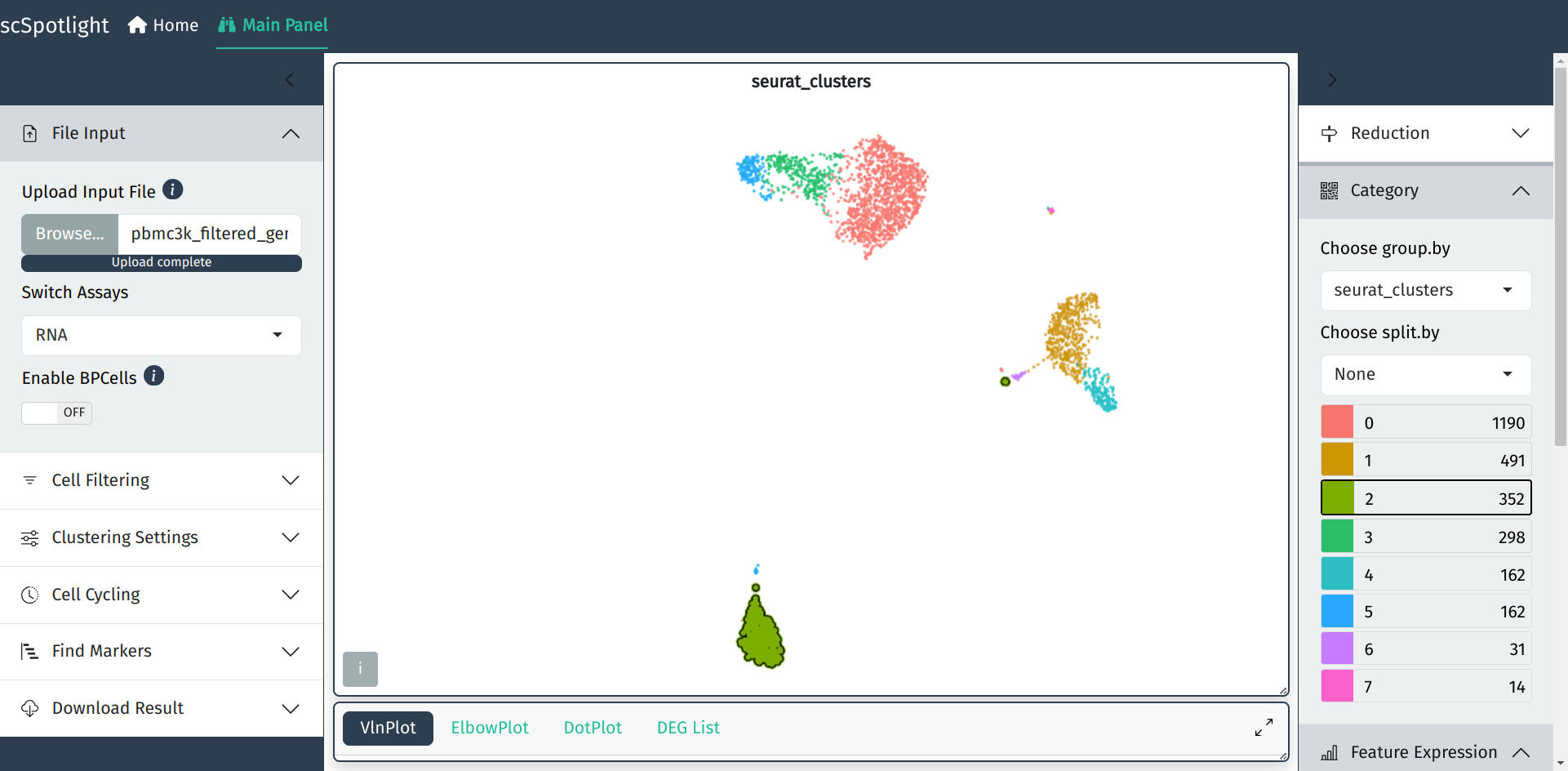
The goal of scSpotlight is to simplify your single cell analysis and easily
annotate your dataset with curated cell type markers. scSpotlight is built on
shiny, Seurat
and regl-scatterplot.
Installation
You can install the development version of scSpotlight from GitHub with:
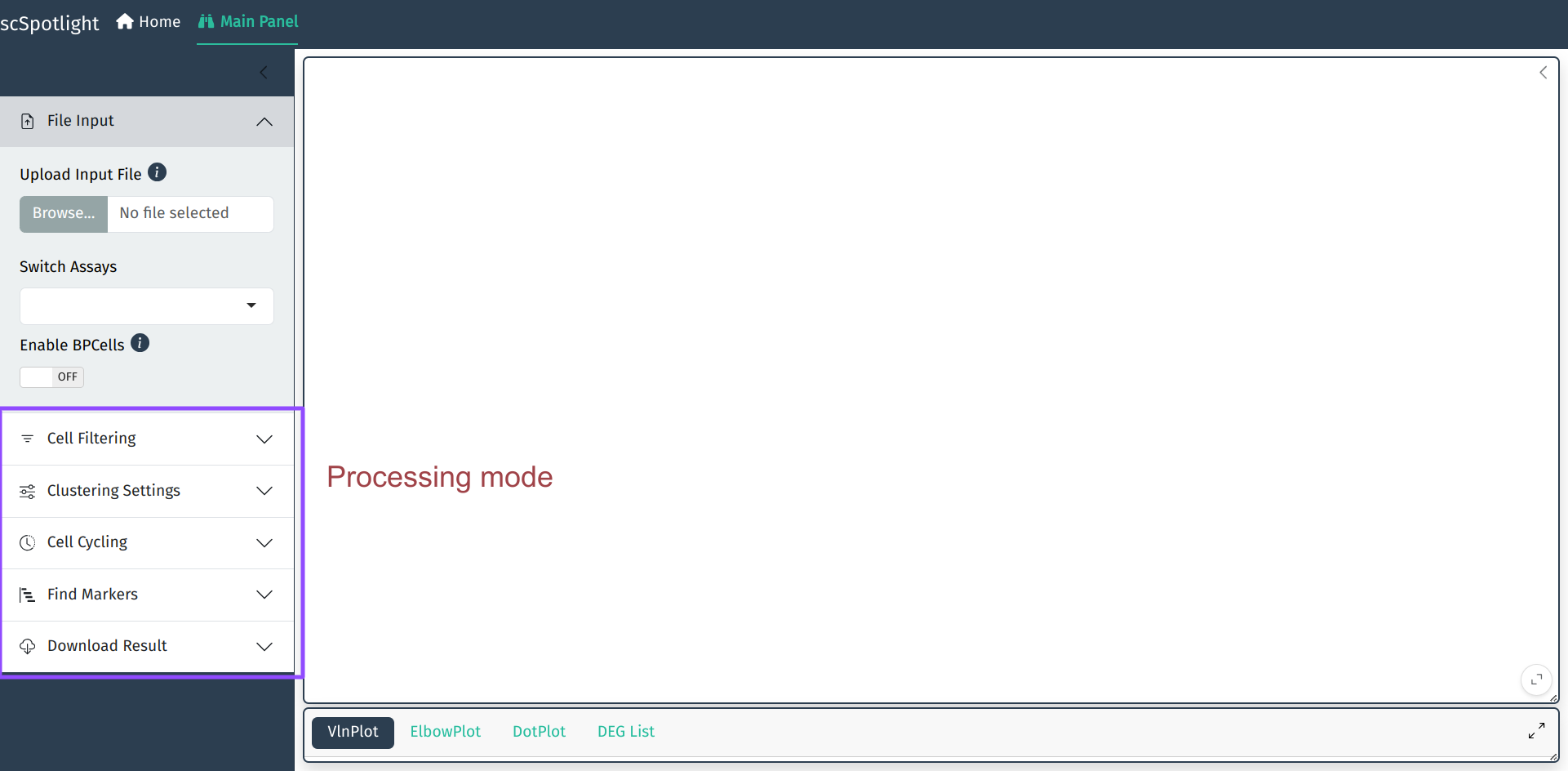
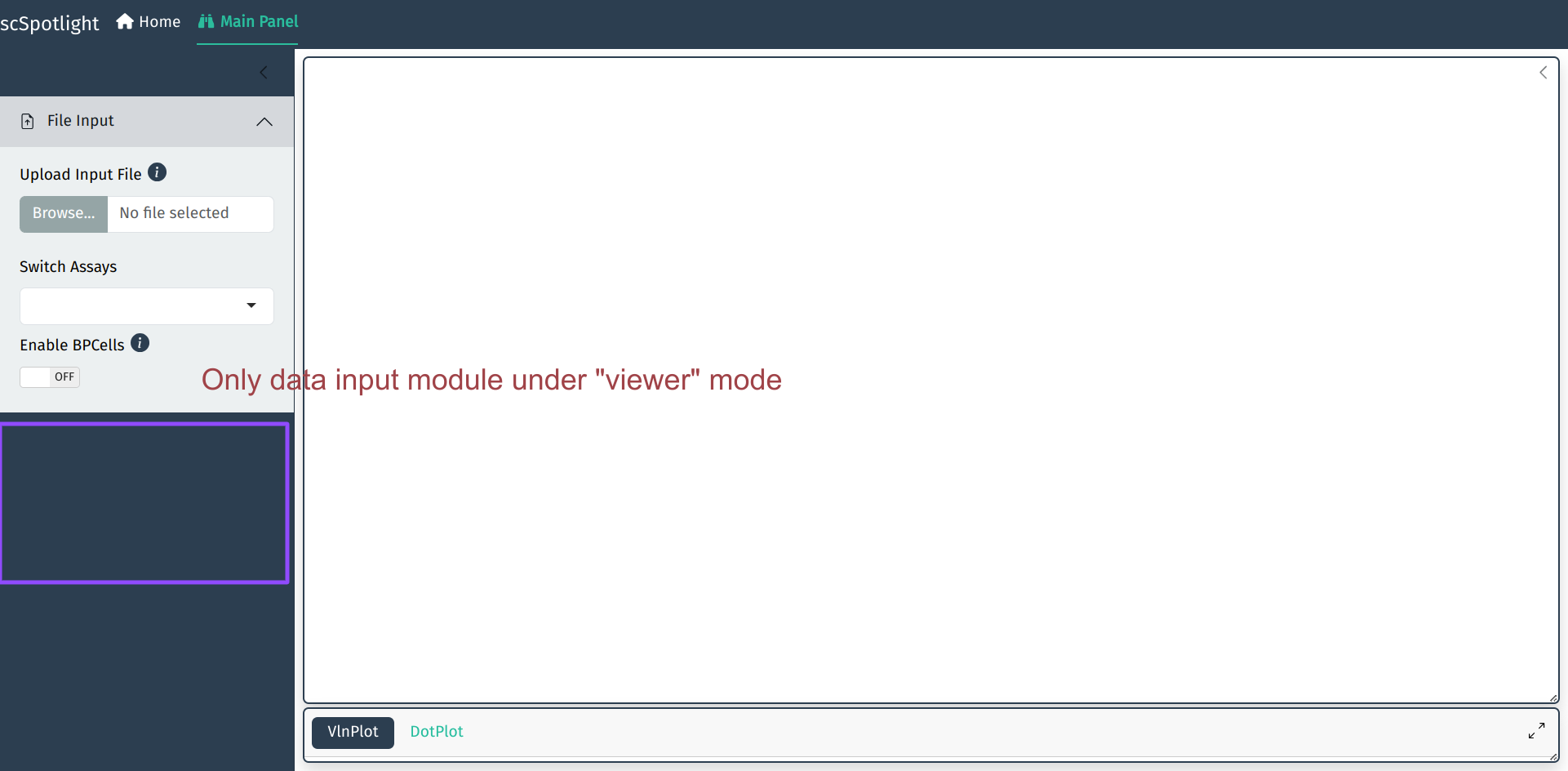
The default mode of app is processing. User could also change mode to viewer, which will only allow illustrating
dataset and querying gene expressions:
After uploading compressed matrix tarball, scSpotlight will
automatically process the data with a standard Seurat workflow
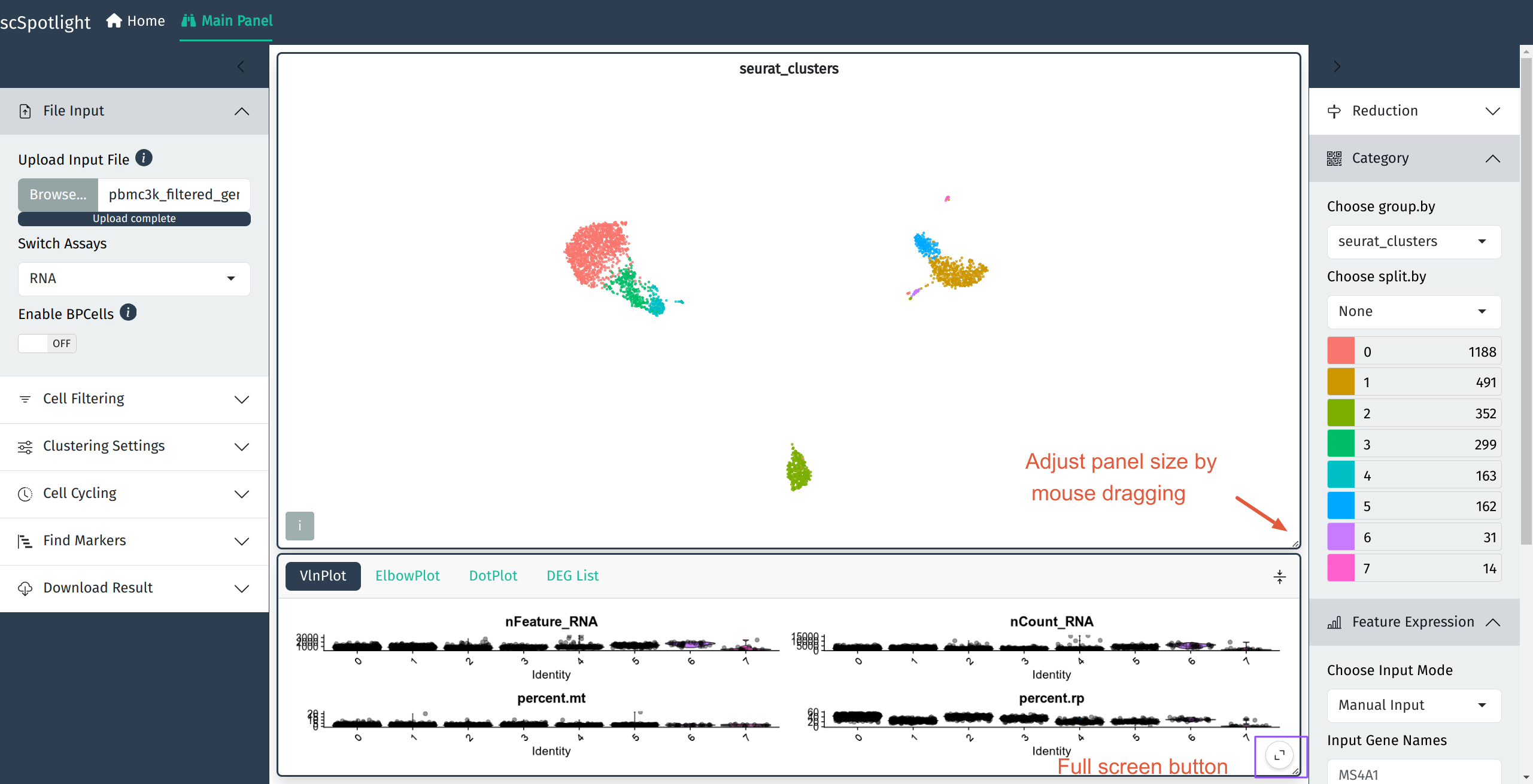
and illustrate the UMAP and cell clusters in the main panel.
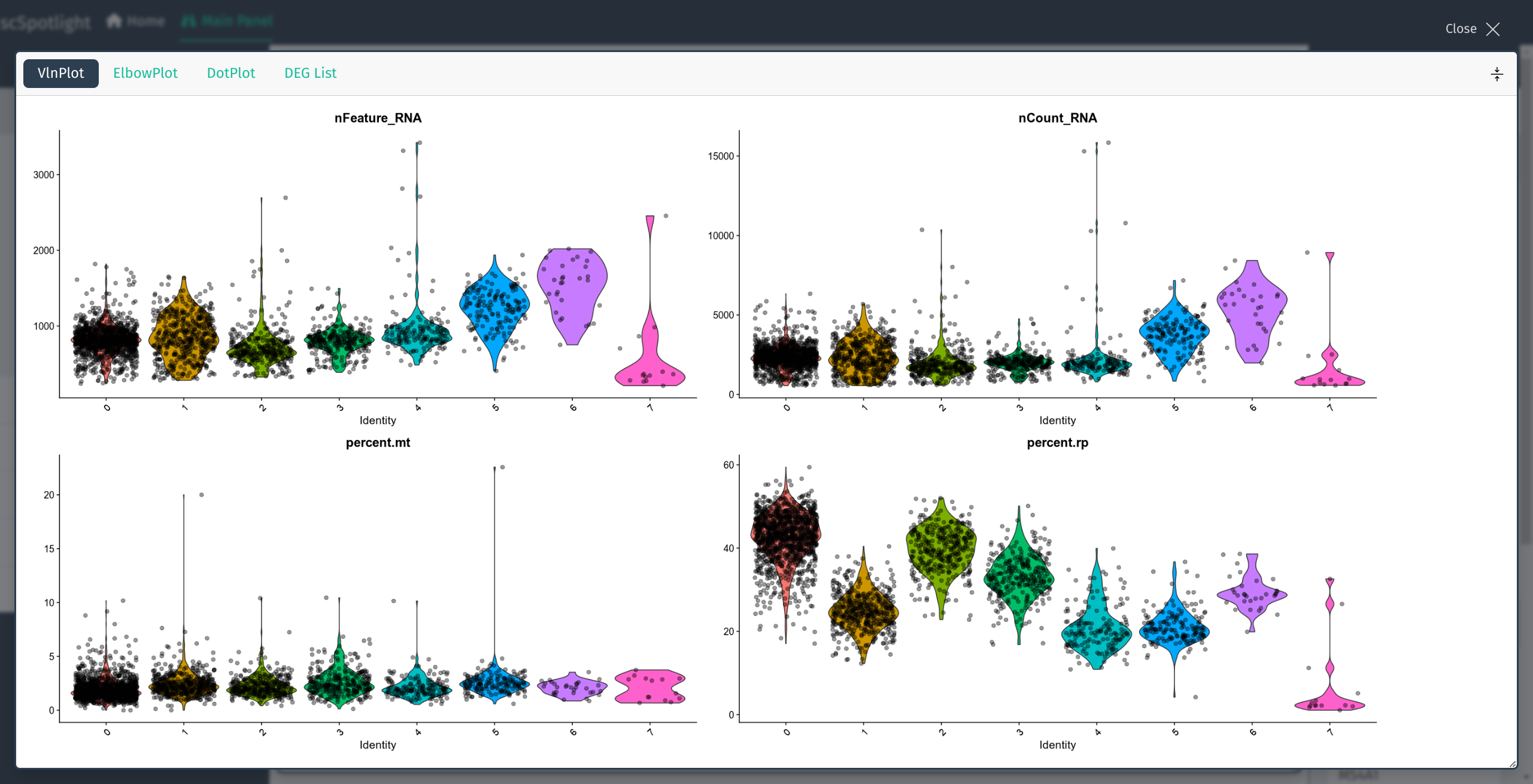
The number of genes detected (nFeature_RNA) in each cell,
number of UMIs (nCount_RNA), percentage of the mitochondrial
UMIs (percent.mt), percentage of the ribosomal protein
UMIs (percent.rp) will be summarized via a violin plot in
the lower information panel.
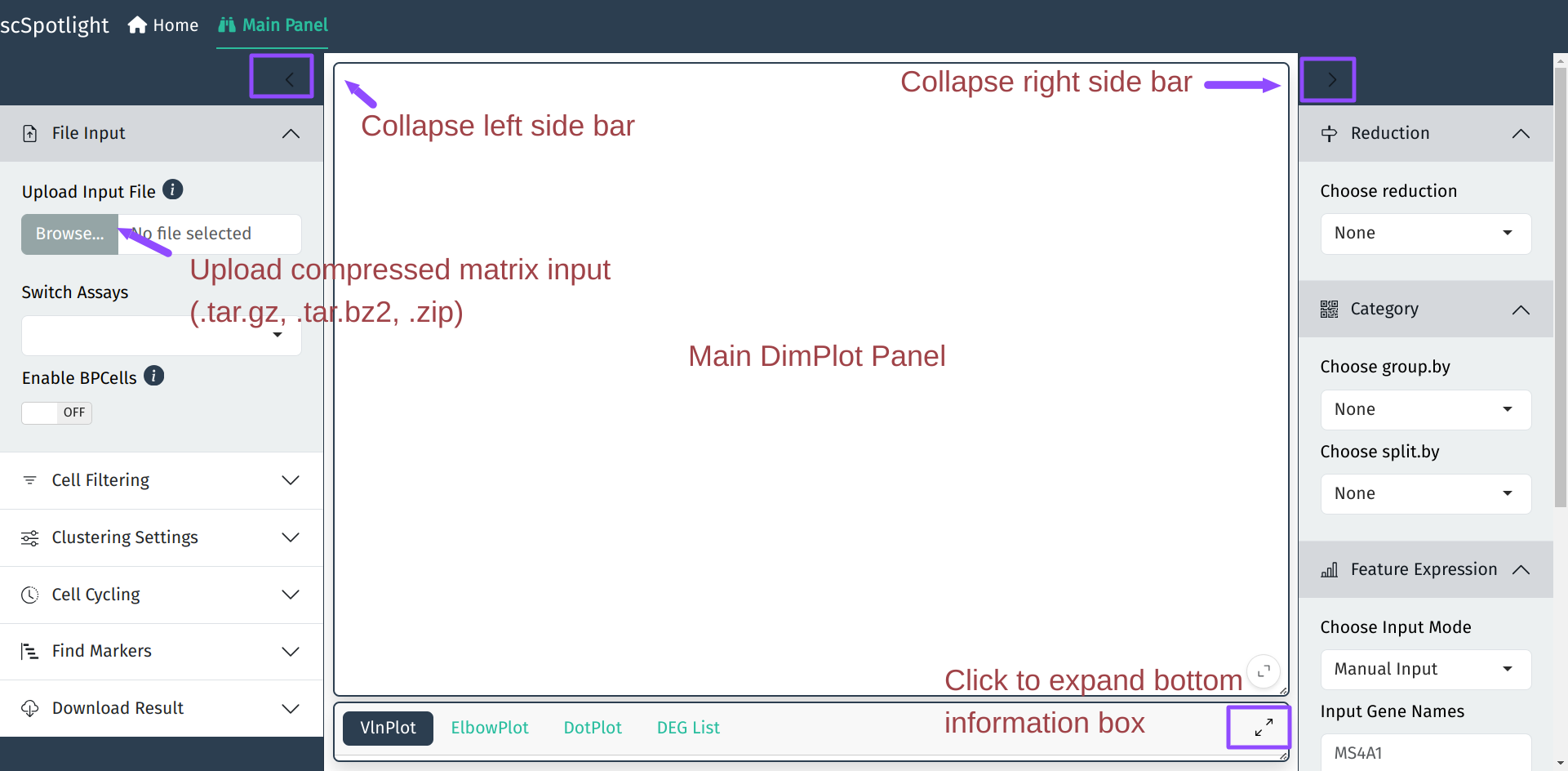
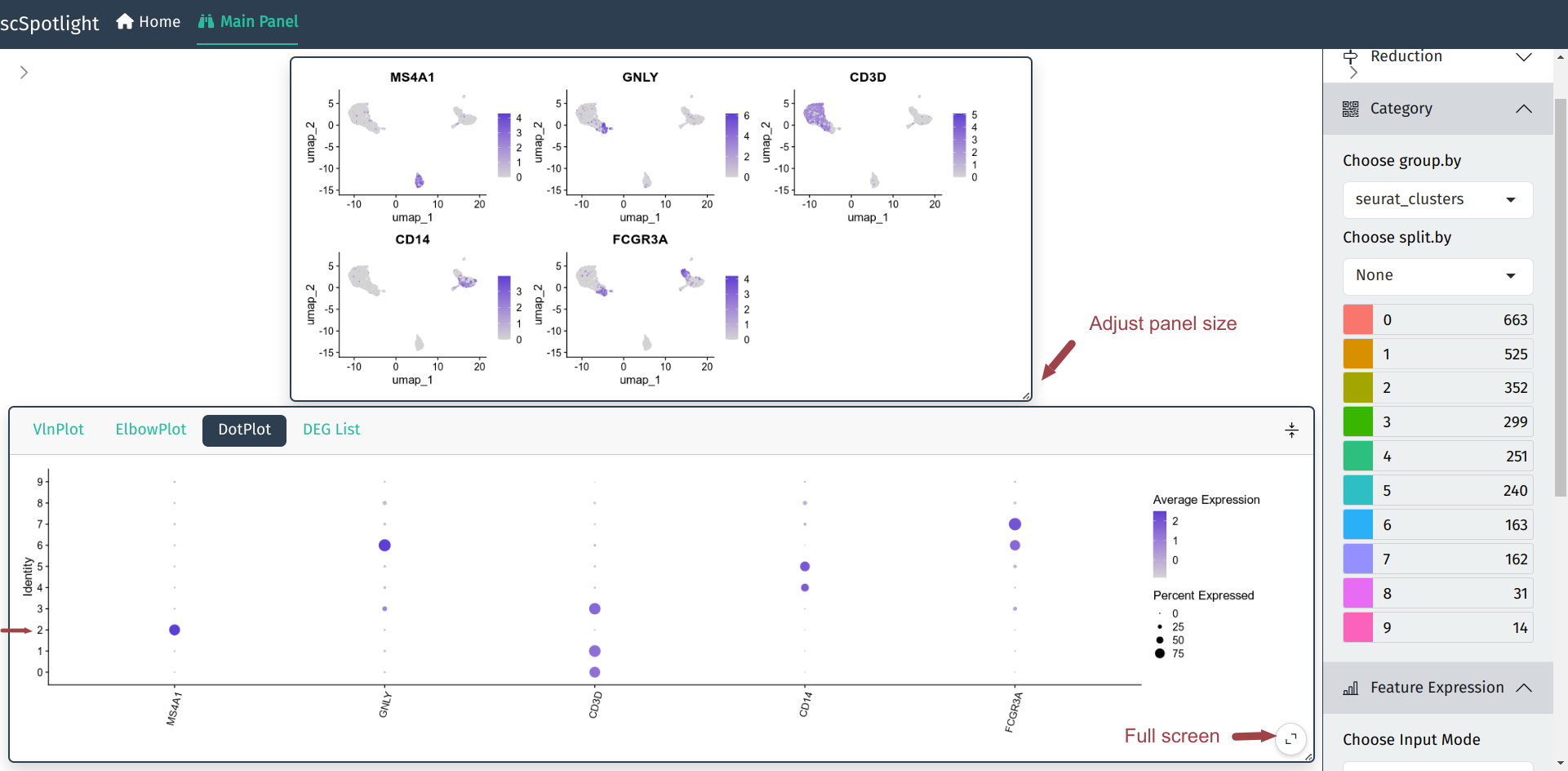
After expanding the informaiton panel, user could drag the
lower right corner of the main panel to adjust the panel size.
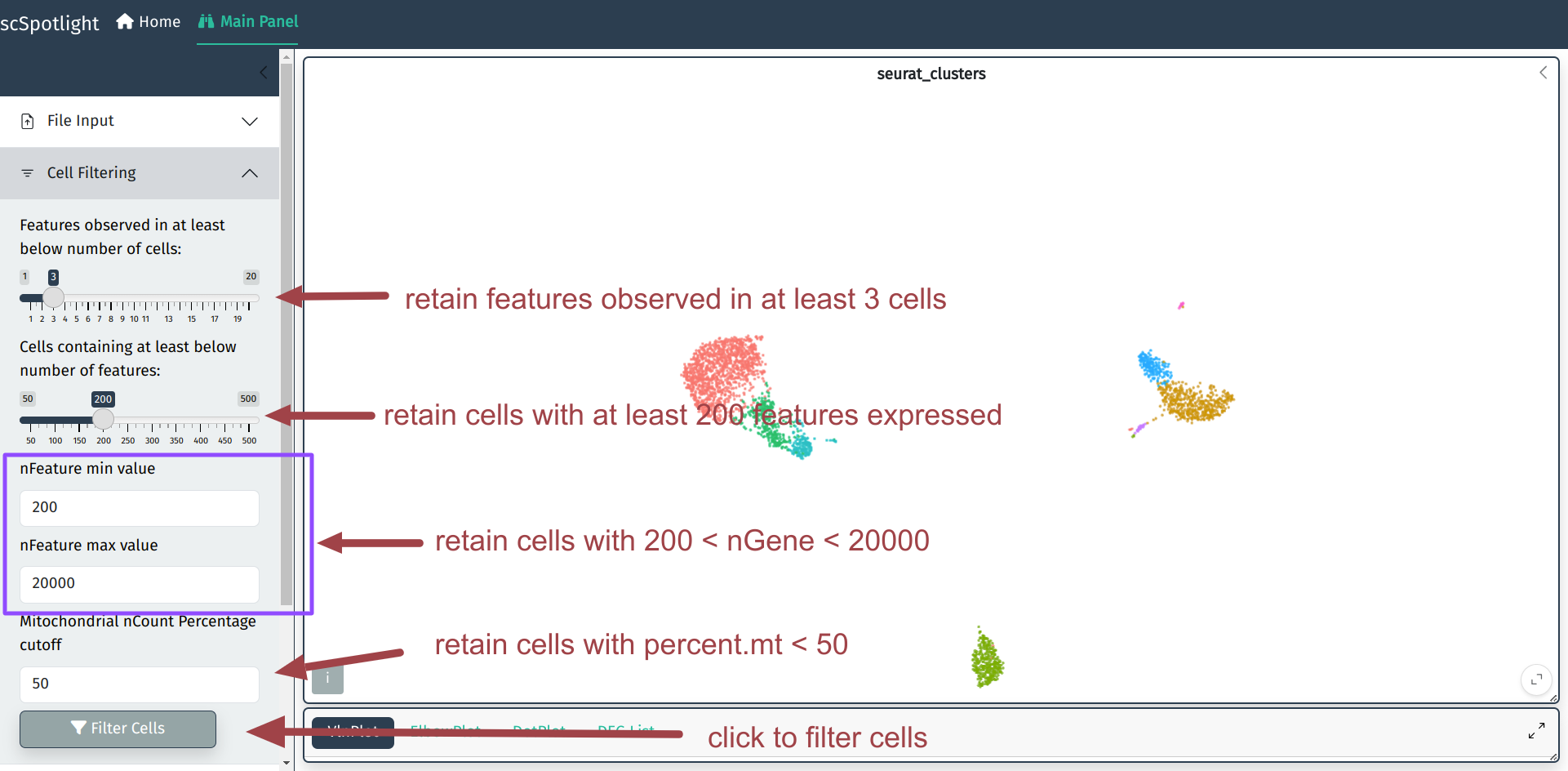
QC metrics and filtration criteria
Here we use the same QC metrics and filtering criteria as Seurat tutorial:
Discard cells/droplets with too few genes detected or too many genes.
Low-quality cells or empty droplets will often have very few genes
Cell doublets or multiplets may exhibit an aberrantly high gene count
Similarly, the total number of molecules detected within a cell (correlates strongly with unique genes), thus only filter nGene is sufficient
Discard cells/droplets with high percentage UMIs belonging to mitochondrial mRNA
Low-quality / dying cells often exhibit extensive mitochondrial contamination
We calculate mitochondrial QC metrics with the PercentageFeatureSet() function, which calculates the percentage of counts originating from a set of features
We use the set of all genes starting with MT- as a set of mitochondrial genes
Discard low quality cells
User could adjust cell filtering criteria, different dataset (with varies sequencing depth)
might need to use different maximum number of genes.
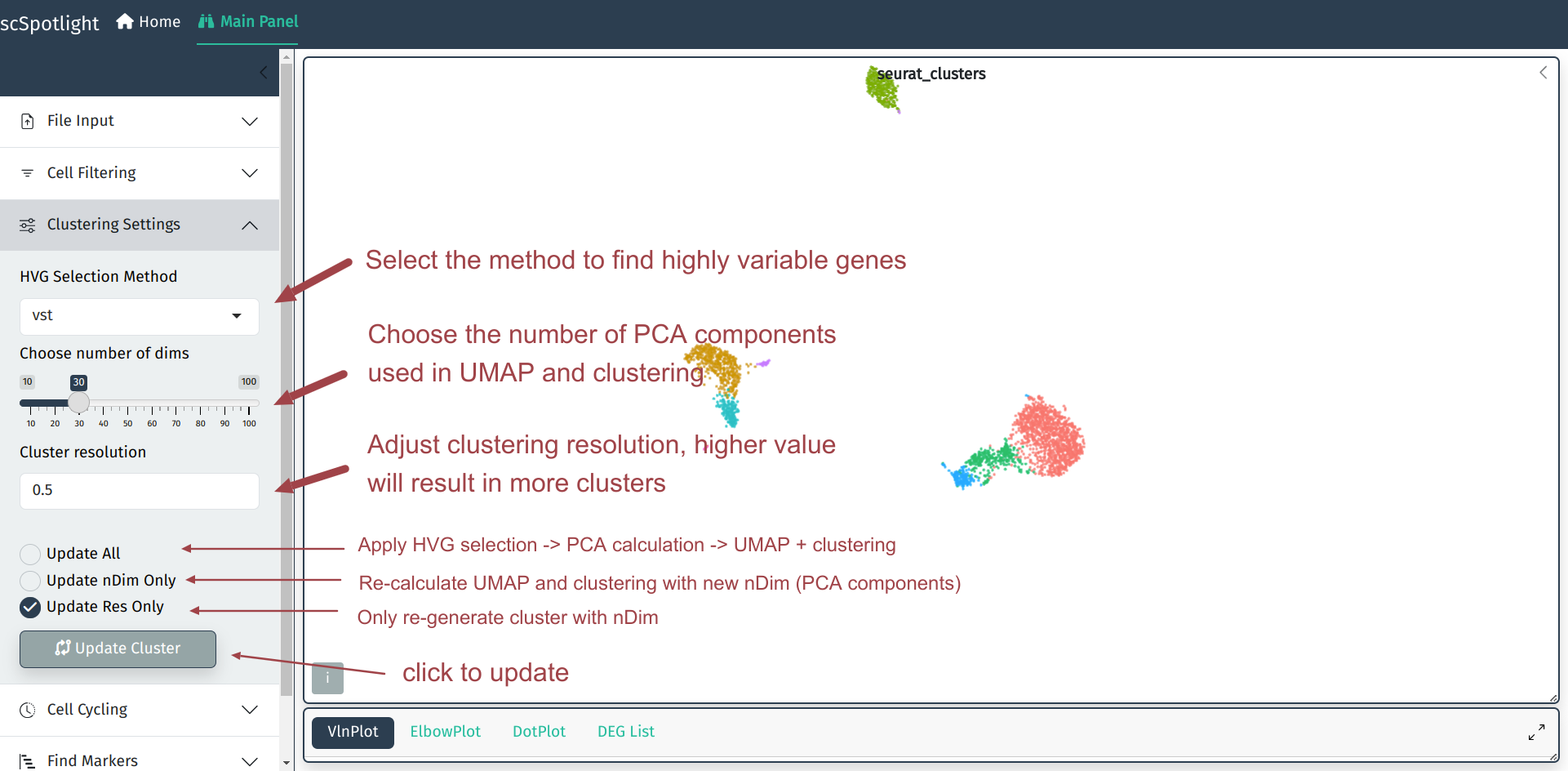
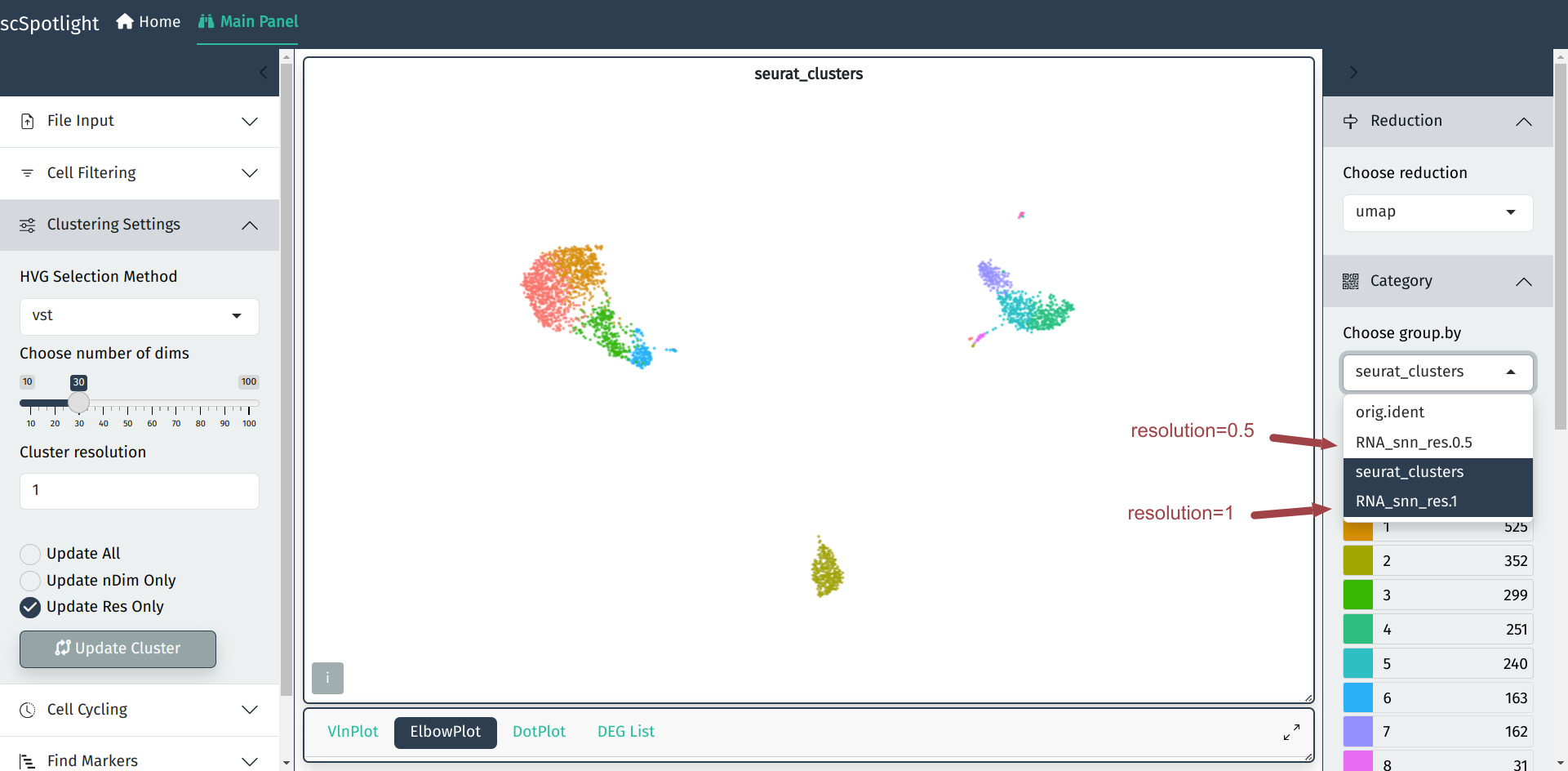
In most cases, the default parameters for clustering will not always
satisfy user’s requirements. scSpotlight allows users to easily choose
the method of highly variable genes selection, tune dimension reduction
and clustering arguments on the left side bar, thus better identify
the rare cell types.
In dimension reduction step, user will have to choose an optimized number of
PCA components as the input of non-linear dimension reduction (UMAP/TSNE).
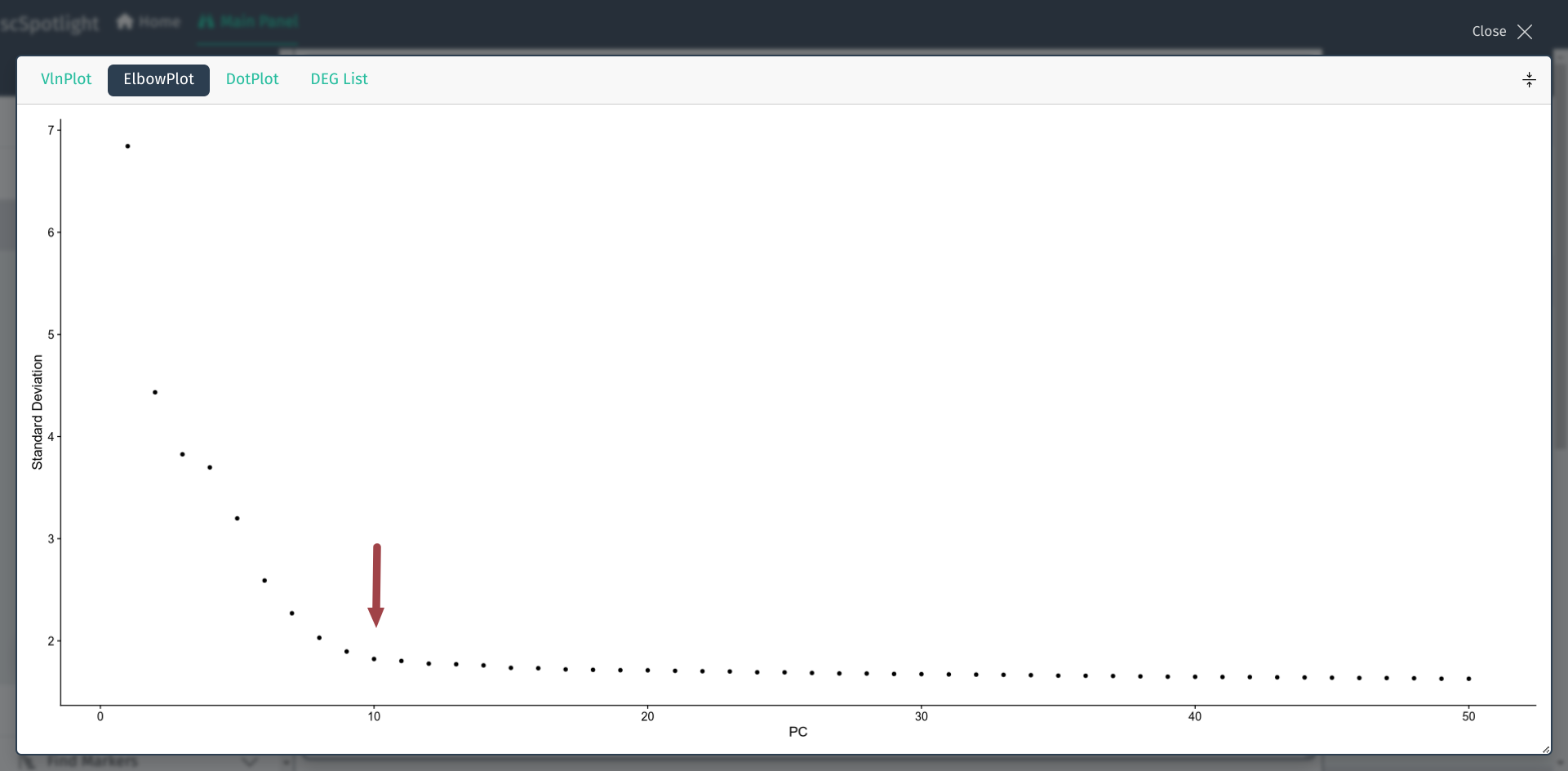
This value could be selected according to elbow plot in Seurat analysis
workflow. User could expand the information panel to view elbow plot, Y axis
is the percentage of the variance that each component could explain,
X axis is the ranked components. Typically, user could select the “elbow”
point. For instance, in Seurat pbmc3k tutorial, they used nDim=10.
After finding cell clusters with different resolution, the updated grouping
information could be chosen from the right-side panel.
The most frequent request during scRNAseq analysis might be checking
genes’ expression, manually annotating cell type is to align
marker gene expression to unsupervised cell clusters. This process
requires user to repeatedly tune the clustering parameters and
verify markers’ expression in the results. scSpotlight tries to
simplify gene query process and make the whole step easier and
more intuitive.
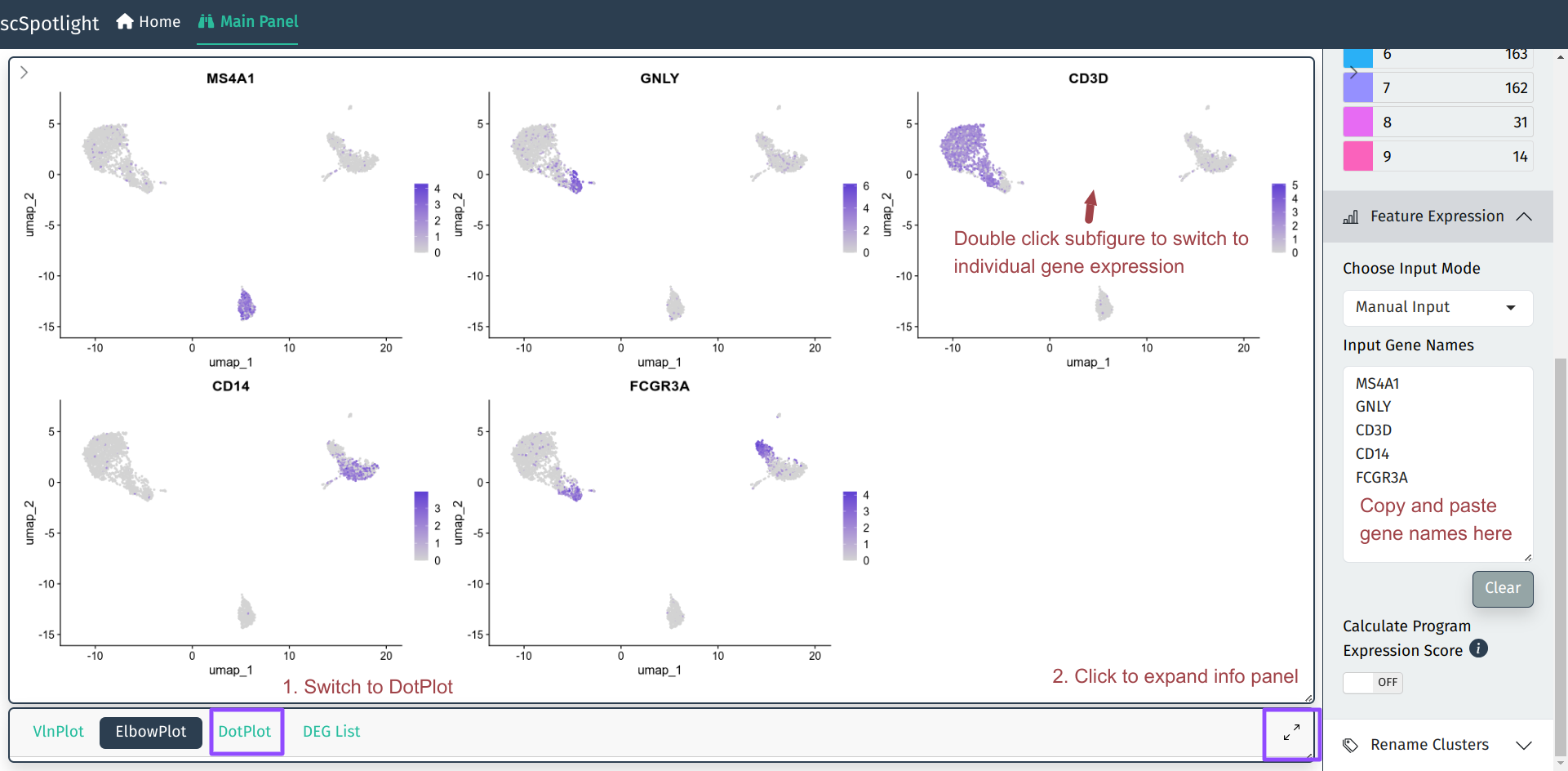
Multiple Genes
User could input multiple gene names (symbols) in the Feature Expression
module and the main plot panel will switch to Seurat::FeaturePlot to
represent gene expression on UMAP plot. User could also check the expression
in Seurat::DotPlot, which could better dipict the percentage of cells
in the cluster expressing the gene of interest.
Individual Gene
For individual genes, user could double click the subfigure of the
FeaturePlot to “zoom in”. The UMP/TSNE of the cell clusters and the
gene expression will be shown side by side. Hovering category
labels will highlight the cells of the cluster. Meanwhile, one also
could view the gene expression in the Seurat::VlnPlot after expanding
the info panel.
scSpotlight allows user to select cells of interest either by category (i.e. group.by)
or manually via the lasso tool. After select cells, user will be able to assign
new identity to them.
Select by category
After choosing “group.by” information from category module, all the
identites of the grouping could be selected from a drop-down menu of
the Rename Cluster module.
For instance, here were illustrated pbmc3k dataset seurat_clusters groups, and overall
expressions of the GNLY gene, which is a “Natrual Kill” (NK) cell marker. We found
cluster 4 cells specifically express GNLY, thus could be assigned to NK cells.
User could select cells of cluster 4 and input a new grouping name (e.g. celltype)
in New Category Name, and assign cell labels “NK” in Assign As.
Unsupervised clustering result is not always perfect, and user may need to
adjust cell annotation manually. One could activate lasso tools by pressing
shift, then click and drag the mouse to select cells. The video
below shows how to select cell of interest with lasso tool and assign new celltype labels.